
Chaque être humain possède plus de 20 000 protéines. Par exemple l’hémoglobine qui s’occupe du transport de l’oxygène depuis les poumons vers les cellules de tout le corps, ou encore l’insuline qui indique à l’organisme la présence de sucre dans le sang.
Chaque protéine est formée d’une suite d’acides aminés, dont la séquence détermine son repliement et sa structure spatiale – un peu comme si un mot se repliait dans l’espace en fonction des enchaînements de lettres dont il est composé. Cette séquence et ce repliement (ou structure) de la protéine déterminent sa fonction biologique : leur étude est le domaine de la « biologie structurale ». Elle s’appuie sur différentes méthodes expérimentales complémentaires, qui ont permis des avancées considérables dans notre compréhension du monde du vivant ces dernières décennies, et permet notamment la conception de nouveaux médicaments.
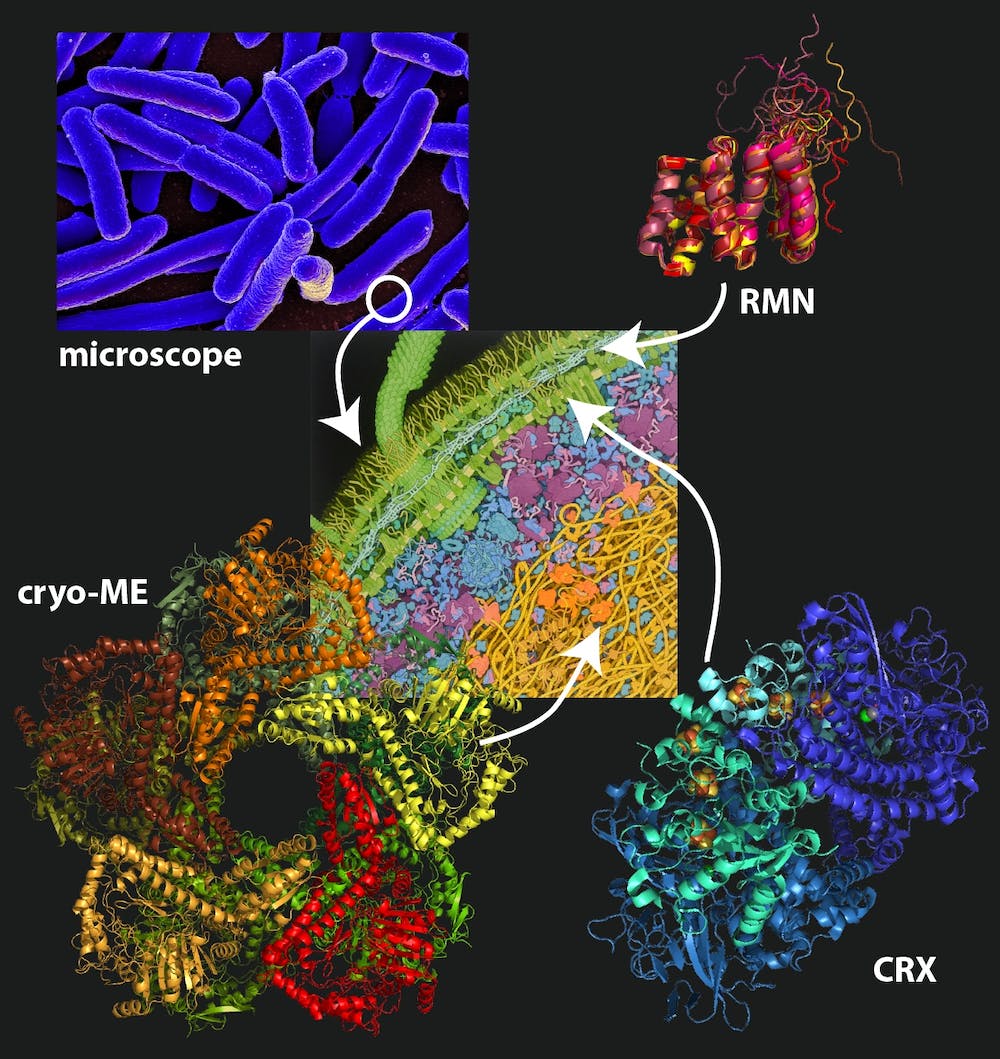
Depuis les années 1970, on cherche à connaître les structures de protéines à partir de la seule connaissance de la séquence d’acides aminés (on dit « ab initio »). Ce n’est que très récemment, en 2020, que ceci est devenu possible de manière quasi systématique, avec l’essor de l’intelligence artificielle et en particulier d’AlphaFold, un système d’IA développé par une entreprise appartenant à Google.
Face à ces progrès de l’intelligence artificielle, quel est désormais le rôle des chercheurs en biologie structurale ?
Pour le comprendre, il faut savoir qu’un des défis de la biologie de demain est la « biologie intégrative », qui a pour objectif de comprendre les processus biologiques au niveau moléculaire dans leurs contextes à l’échelle de la cellule. Vu la complexité des processus biologiques, une approche pluridisciplinaire est indispensable. Elle s’appuie sur les techniques expérimentales, qui restent incontournables pour l’étude de la structure des protéines, leur dynamique et leurs interactions. De plus, chacune des techniques expérimentales peut bénéficier à sa manière des prédictions théoriques d’AlphaFold.
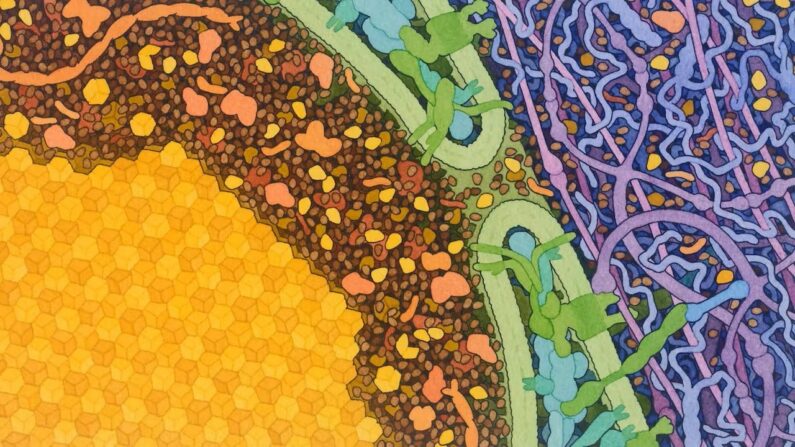
Beate Bersch, IBS, à partir d’une illustration de David Goodsell, Fourni par l’auteur
La cristallographie aux rayons X
La cristallographie est, à cette date, la technique la plus utilisée en biologie structurale. Elle a permis de recenser plus de 170 000 structures de protéines dans la « Protein Data Bank », avec plus de 10 000 repliements différents.
Pour utiliser la cristallographie à rayons X, il faut faire « cristalliser les protéines ». On dit souvent que cette technique est limitée par la qualité de cristaux de protéines, qui est moindre pour les grosses protéines. Mais cette notion ne correspond pas toujours à la réalité : par exemple, la structure du ribosome, l’énorme machine moléculaire qui assemble les protéines, a été résolue à 2,8 angströms de résolution. Venkatraman Ramakrishnan, Thomas Steitz et Ada Yonath ont reçu le prix Nobel de chimie en 2009 pour ce travail.
Avec le développement récent du laser X à électron libre (XFEL), il est devenu possible d’étudier simultanément des milliers de microcristaux de protéines à température ambiante et à l’échelle de la femtoseconde (10-15 secondes, soit un millionième de milliardième de seconde, l’échelle de temps à laquelle ont lieu les réactions chimiques et le repliement des protéines). Cette technique permet d’imager les protéines avant qu’elles ne soient détruites. Elle est en train de révolutionner la « cristallographie cinétique », qui permet de voir les protéines « en action », ainsi que la recherche de médicaments.
Pour l’instant, l’apport d’AlphaFold à l’étude de la structure des protéines par cristallographie s’est concentré dans la génération de modèles de protéines assez précis pour appliquer la technique dite de « remplacement moléculaire » à la résolution des structures.
La spectroscopie par résonance magnétique nucléaire
Une autre méthode expérimentale pour étudier la structure des protéines est la « spectroscopie par résonance magnétique nucléaire ». Alors que son alter ego d’imagerie médicale, l’IRM, regarde la distribution spatiale d’un seul signal, caractéristique des éléments chimiques dans les tissus biologiques observés, en spectroscopie par résonance magnétique nucléaire, c’est un ensemble de signaux provenant des atomes constituant la protéine qui est enregistré (ce qu’on appelle le « spectre »).
Généralement, la détermination de la structure par résonance magnétique est limitée à des protéines de taille modeste. On calcule des modèles de molécules basés sur des paramètres structuraux (comme des distances interatomiques), provenant de l’analyse des spectres expérimentaux. On peut s’imaginer cela comme dans les débuts de la cartographie, où des distances entre des points de référence permettaient de dessiner des cartes en 2D. Pour faciliter l’interprétation des spectres qui contiennent beaucoup d’information, on peut utiliser des modèles obtenus par prédiction (plutôt qu’expérimentalement), comme avec AlphaFold.
En plus de la détermination structurale, la spectroscopie par résonance magnétique nucléaire apporte deux atouts majeurs. D’une part, en général, l’étude est effectuée avec un échantillon en solution aqueuse et il est possible d’observer les parties particulièrement flexibles des protéines, souvent invisibles avec les autres techniques. On peut même quantifier leur mouvement en termes d’amplitude et de fréquence, ce qui est extrêmement utile car la dynamique interne des protéines est aussi cruciale pour leur fonctionnement que leur structure.
D’autre part, la spectroscopie par résonance magnétique nucléaire permet de détecter aisément les interactions des protéines avec des petites molécules (ligands, inhibiteurs) ou d’autres protéines. Ceci permet d’identifier les sites d’interaction, information essentielle entre autres pour la conception rationnelle de molécules actives comme des médicaments.
Ces propriétés font de la spectroscopie par résonance magnétique nucléaire un outil extraordinaire pour la caractérisation fonctionnelle des protéines en complémentarité avec d’autres techniques expérimentales et l’IA.
La « cryomicroscopie électronique »
La cryomicroscopie électronique consiste à congeler ultrarapidement (environ -180 °C) un échantillon hydraté dans une fine couche de glace, qui sera traversée par les électrons. Les électrons transmis vont générer une image de l’échantillon, qui après analyse, permet d’accéder à des structures qui peuvent atteindre la résolution atomique. En comparaison, un microscope optique n’a un pouvoir résolutif que de quelques centaines de nanomètres, qui correspond à la longueur d’onde de la lumière utilisée ; seul un microscope utilisant une source possédant des longueurs d’onde suffisamment faibles (comme les électrons pour la microscopie électronique) possède un pouvoir résolutif théorique de l’ordre de l’angström. Le prix Nobel de Chimie 2017 a été décerné à Jacques Dubochet, Richard Henderson et Joachim Frank pour leurs contributions au développement de la cryomicroscopie électronique.
Avec de nombreux développements technologiques, dont celui des détecteurs à électrons directs, depuis le milieu des années 2010, cette technique est devenue essentielle en biologie structurale en amorçant une « révolution de la résolution ». En effet, la cryomicroscopie électronique permet désormais d’obtenir des structures avec une résolution atomique, comme dans le cas de l’apoferritine – une protéine de l’intestin grêle qui contribue à l’absorption du fer – à 1,25 angström de résolution.
Son principal atout est de permettre de déterminer la structure d’objets de taille moyenne, au-delà de 50 000 Dalton (un Dalton correspond environ à la masse d’un atome d’hydrogène), comme l’hémoglobine de 64 000 Dalton, mais également d’objets de quelques milliards de daltons (comme le mimivirus, virus géant d’environ 0,5 micromètre).
Malgré toutes les avancées technologiques précédemment évoquées, la cryomicroscopie ne permet pas toujours de résoudre à suffisamment haute résolution la structure de « complexes », constitués de plusieurs protéines. C’est ici qu’AlphaFold peut aider et permettre, en complémentarité avec la cryomicroscopie, de décrire les interactions au niveau atomique entre les différents constituants d’un complexe. Cette complémentarité donne une force nouvelle à la cryomicroscopie électronique pour son rôle à jouer demain en biologie structurale.
Les apports d’AlphaFold
AlphaFold permet de prédire la structure de protéines uniquement à partir de leur séquence avec la connaissance acquise par la biologie structurale expérimentale. Cette approche est révolutionnaire car les séquences de beaucoup de protéines sont connues à travers les efforts des séquençages des génomes, mais déterminer leurs structures expérimentalement nécessiterait des moyens humains et techniques colossaux.
À l’heure actuelle, ce type de programme représente donc un acteur supplémentaire de complémentarité, mais ne se substitue pas aux techniques expérimentales qui, comme nous l’avons vu, apportent aussi des informations complémentaires (dynamiques, interfaces), à des échelles différentes (des sites métalliques aux complexes multiprotéiques) et plus fiables, car expérimentalement vérifiées. Au-delà de la pure détermination structurale d’une protéine isolée, la complexité des systèmes biologiques nécessite souvent une approche pluridisciplinaire afin d’élucider mécanismes et fonctions de ces biomolécules fascinantes que sont les protéines.![]()
Beate Bersch, Chercheuse CNRS à l’Institut de Biologie Structurale, Université Grenoble Alpes (UGA); Emmanuelle Neumann, Engineer-Researcher CEA, Université Grenoble Alpes (UGA) et Juan Fontecilla, Chercheur en biologie structurale, Université Grenoble Alpes (UGA)
Cet article est republié à partir de The Conversation sous licence Creative Commons. Lire l’article original.
Comment pouvez-vous nous aider à vous tenir informés ?
Epoch Times est un média libre et indépendant, ne recevant aucune aide publique et n’appartenant à aucun parti politique ou groupe financier. Depuis notre création, nous faisons face à des attaques déloyales pour faire taire nos informations portant notamment sur les questions de droits de l'homme en Chine. C'est pourquoi, nous comptons sur votre soutien pour défendre notre journalisme indépendant et pour continuer, grâce à vous, à faire connaître la vérité.












