
La pandémie liée à SARS-CoV-2 pose de façon aiguë la question du design de molécules capables de limiter l’action d’un virus sur nos cellules – un mécanisme qui implique des molécules de très grande taille difficiles à modéliser, des protéines, qui sont de plus… en mouvement permanent.
Des systèmes d’intelligence artificielle, à commencer par AlphaFold2 de Google, prédisent désormais la configuration de ces protéines de façon impressionnante, ce qui révolutionne la recherche dans le domaine. Comment marchent ces méthodes ? Quelles sont leurs limites actuelles ?
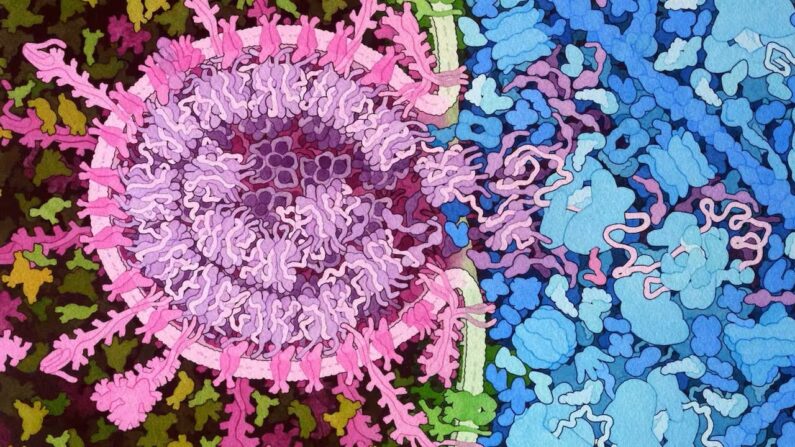
Entrée de SARS-CoV-2 : une histoire de monte-en-l’air
L’infection de l’une de nos cellules par le SARS-CoV-2, le virus responsable du Covid-19, commence par une sorte d’effraction : le virus, une enveloppe hérissée de protéines à l’intérieur de laquelle se trouve son matériel génétique, se comporte comme un voleur entrant dans un appartement au premier étage d’un immeuble. Avec un grappin (le « domaine de fixation au récepteur » ou RBD se trouvant sur la fameuse protéine « spike »), il s’accroche à la rambarde (la non moins fameuse protéine « ACE2 »). Puis, à l’aide d’un marteau (le domaine de fusion, une autre région du spike), il brise la vitre (la membrane de la cellule) et injecte son matériel génétique.
Ce mécanisme est dynamique, c’est-à-dire que les molécules changent de conformation (de forme) pendant l’effraction. D’une part, le virus ne « dégaine » son grappin qu’au dernier instant ; d’autre part, le « bris de vitre » utilise une sorte de perche télescopique dont l’assemblage est complexe.
Ces deux protagonistes (spike et ACE2) sont des protéines. Les interactions entre protéines sont à la base de l’immense majorité des fonctions biologiques, et la compréhension de ces interactions nécessite dans un premier temps la connaissance de la forme géométrique des partenaires – on parle souvent de « clef » et de « serrure » pour visualiser le fait que la géométrie des protéines doit être adéquate pour qu’elles interagissent.
Ces conformations moléculaires sont étudiées expérimentalement depuis les années 1950-60 et entreposées dans une base de données internationale, la Protein Data Bank.
Dans le cas du SARS-CoV-2, on a beaucoup présenté la protéine Spike comme une telle clef, qui s’emboîterait dans la « serrure » ACE2. Mais le mécanisme clef-serrure est une vision un peu simpliste, et comme on l’a vu les protéines sont dotées d’une certaine flexibilité (elles se déforment), ce qui leur permet également de s’adapter.
En effet, une façon de bloquer l’infection par le SARS-CoV-2 consiste à empêcher la fixation du grappin (la protéine Spike), et plus précisément de son domaine de fixation au récepteur (RBD) sur la cible ACE2. C’est l’objectif de certains anticorps sécrétés par notre système immunitaire.
Hélas, par le jeu des mutations, le virus cherche en permanence à échapper à ce contrôle : certains acides aminés changent, ce qui fait que la conformation de sa protéine spike n’est plus reconnue par les anticorps. Ceux-ci n’ayant plus une affinité suffisante, le système immunitaire doit s’adapter, ce qui est une gageure lorsqu’il s’agit d’être efficace face à un large éventail de souches virales.
Affinité entre deux biomolécules : structure et dynamique
Pour mieux comprendre la fixation du « grappin » (RBD) sur la « rambarde » (ACE2), intéressons-nous à l’interaction deux protéines A et B formant un complexe C.
À l’échelle atomique, deux phénomènes sont en compétition : des forces d’attraction entre atomes font que les molécules s’attirent ; mais, sous l’effet de l’agitation thermique – c’est-à-dire les déplacements aléatoires des atomes qui augmentent avec la température, les molécules se déforment.
Cette agitation thermique fait qu’une fois le complexe C formé, il peut se dissocier en A et B, les partenaires pouvant alors s’associer à nouveau, et ainsi de suite. Il s’agit là d’un équilibre chimique, et la quantité relative de molécules A et B et du complexe C permet de mesurer la stabilité de l’interaction. Plus il y a de complexe C, plus cela signifie que l’affinité de A pour B est élevée, et donc que leur interaction est stable.
Dans le cas de Spike et ACE2, une haute affinité du « grappin » (RBD) pour la « rambarde » (ACE2) augmentera le pouvoir infectieux du virus (le grappin s’accrochera d’autant plus fortement à la rambarde que son affinité pour elle est grande).
AlphaFold2 : de la structure à l’affinité
Estimer l’affinité de liaison nécessite donc de prendre en compte les déformations autour d’une structure moléculaire moyenne. Dans la métaphore serrure-clef, la forme de cette dernière doit être connue, au moins approximativement. On sait que les protéines sont formées de longues chaînes de différents acides aminés attachés ensemble, comme un long collier de perles.
Connaissant la séquence des acides aminés d’une protéine (autrement dit, l’ordre dans lequel ils s’enchaînent), pourrait-on prédire la forme qu’elle adoptera, en la calculant par ordinateur ?
Ce sujet a fait l’objet d’une avancée majeure avec le développement de la méthode AlphaFold2 et du logiciel éponyme, par un groupe de recherche de Google DeepMind. Cette méthode a très clairement surpassé ses concurrentes lors du concours CASP14 en 2020, qui évalue la qualité des prédictions en les comparant à des structures résolues expérimentalement mais non révélées aux compétiteurs.
De façon très schématique, étant donnée la séquence d’acides aminés dont la conformation doit être prédite, AlphaFold2 utilise en entrée une base de données de séquences homologues (séquences différentes mais pour lesquelles les changements d’acides aminés n’altèrent pas la fonction de la protéine), ainsi que certaines structures expérimentales issues de la Protein Data Bank. La méthode génère en sortie une structure plausible pour la protéine, ainsi qu’un « score de confiance » pour la position, une fois la protéine repliée, de chaque acide aminé dans la conformation calculée, ce qui permet de voir quels acides aminés sont exposés et peuvent interagir avec l’extérieur.
La méthode utilise deux blocs principaux. Le premier produit un modèle grossier codant certaines contraintes entre les acides aminés, notamment les distances trois à trois qui doivent respecter l’inégalité triangulaire. Le second, le module de structure, introduit explicitement le modèle 3D en positionnant les acides aminés les uns par rapport aux autres, grâce à des « mécanismes d’attention », une technique algorithmique permettant d’explorer des hypothèses de façon aléatoire, et de retenir celles qui sont les plus cohérentes avec le modèle en cours d’élaboration. In fine, le réseau de neurones génère une conformation plausible.
À ce jour, la méthode est particulièrement efficace pour des domaines de protéines bien structurés (les plus rigides), mais l’est beaucoup moins pour les parties non structurées (les plus flexibles), ou encore pour les boucles flexibles pour lesquelles la notion de même structure unique n’a pas de sens. Par ailleurs, en dépit du score de confiance évoqué ci-dessus, le résultat global est livré sans garantie aucune.
Appliquer la méthode aux anticorps du SARS-CoV-2
Le succès tonitruant de cette méthode a bien entendu éveillé un intérêt pour la prédiction d’affinité, qui a été explorée très récemment pour optimiser des anticorps contre le RDB du SARS- Cov-2, afin que ces anticorps aient une affinité élevée pour des souches virales différentes.
La méthode utilise pour cela une base de données de « mutagenèse » : celle-ci donne à la fois la structure d’un complexe, la structure d’un complexe analogue dont les protéines ont muté génétiquement, et également l’affinité associée à chacun de ces deux complexes. Il s’agit donc d’apprendre comment les mutations influencent l’affinité. D’un point de vue méthodologique, l’algorithme identifie les acides aminés contribuant significativement à l’affinité de liaison.
De façon remarquable, cette stratégie a permis d’optimiser un anticorps efficace contre les variants Alpha, Beta et Gamma du SARS-CoV-2 (mais pas Delta).
La prédiction de la dynamique reste un problème ouvert
Estimer de façon fiable l’affinité de liaison entre des grosses molécules comme les protéines nécessite d’explorer des espaces de très haute dimension (les atomes sont nombreux et bougent dans les 3 dimensions de l’espace) afin de calculer les propriétés moyennes rendant compte de nos observations macroscopiques.
De plus, dans le contexte d’AlphaFold2 et de l’apprentissage machine, il faut des données disponibles, afin que les algorithmes puissent apprendre à lier la structure et ses propriétés. Dans notre cas, les informations statiques présentes dans la Protein Data Bank et autres bases de données ne contiennent manifestement pas toute l’information dynamique requise.
« Prédire n’est pas expliquer »
La question pratique du blocage effectif d’un virus comme SARS-CoV-2 montre à quel point ces questions de design moléculaire sont difficiles, ne relevant pas à ce jour d’un travail d’optimisation d’ingénierie classique.
La prédiction d’affinité illustre également l’opposition observée en épistémologie entre « prédictivisme » et explication par des lois et modèles, qui permettent d’établir une chaîne de causalité. Comme le disait le mathématicien et épistémologue René Thom, « Prédire n’est pas expliquer », et les techniques d’apprentissage machine illustrent bien cette dissonance.
Gageons cependant que l’accumulation de données, dynamiques en particulier, permettra une convergence dans le sens où l’apprentissage machine sera capable d’assortir ses prédictions d’explications.![]()
Frédéric Cazals, Directeur de recherche; Equipe Algorithms-Biology-Structure, Inria
Cet article est republié à partir de The Conversation sous licence Creative Commons. Lire l’article original.
Comment pouvez-vous nous aider à vous tenir informés ?
Epoch Times est un média libre et indépendant, ne recevant aucune aide publique et n’appartenant à aucun parti politique ou groupe financier. Depuis notre création, nous faisons face à des attaques déloyales pour faire taire nos informations portant notamment sur les questions de droits de l'homme en Chine. C'est pourquoi, nous comptons sur votre soutien pour défendre notre journalisme indépendant et pour continuer, grâce à vous, à faire connaître la vérité.












